Cystisk fibros

Cystisk fibros är en sjukdom som får tjockt, klibbigt slem att byggas upp i lungorna, mag-tarmkanalen och andra delar av kroppen. Det är en av de vanligaste kroniska lungsjukdomarna hos barn och unga vuxna. Det är en livshotande sjukdom.
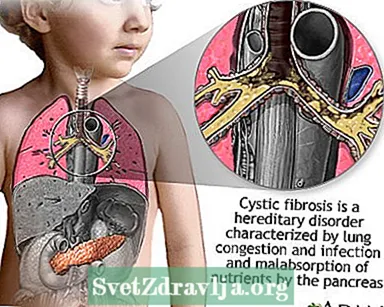
Cystisk fibros (CF) är en sjukdom som överförs genom familjer. Det orsakas av en defekt gen som gör att kroppen producerar onormalt tjock och klibbig vätska, kallad slem. Detta slem byggs upp i lungorna och i bukspottkörteln.
Uppbyggnaden av slem resulterar i livshotande lunginfektioner och allvarliga matsmältningsproblem. Sjukdomen kan också påverka svettkörtlarna och en mans reproduktionssystem.
Många har en CF-gen men har inga symtom. Detta beror på att en person med CF måste ärva 2 defekta gener, 1 från varje förälder. Vissa amerikaner har CF-genen. Det är vanligare bland de av norra eller centraleuropeiska härkomst.
De flesta barn med CF diagnostiseras vid 2 års ålder, särskilt när nyfödda screening utförs över hela USA. För ett litet antal upptäcks sjukdomen inte förrän 18 år eller äldre. Dessa barn har ofta en mildare form av sjukdomen.
Symtom hos nyfödda kan inkludera:
- Försenad tillväxt
- Underlåtenhet att gå upp i vikt normalt under barndomen
- Inga tarmrörelser under de första 24 till 48 timmarna av livet
- Salt smakande hud
Symtom relaterade till tarmfunktionen kan inkludera:
- Magbesvär från svår förstoppning
- Ökad gas, uppblåsthet eller mage som verkar svullen (utspänd)
- Illamående och aptitlöshet
- Avföring som är blek eller lerfärgad, illaluktande, har slem eller som flyter
- Viktminskning
Symtom relaterade till lungor och bihålor kan inkludera:
- Hosta eller ökat slem i bihålorna eller lungorna
- Trötthet
- Trängsel i näsan orsakad av näspolyper
- Upprepade episoder av lunginflammation (symtom på lunginflammation hos någon med cystisk fibros inkluderar feber, ökad hosta och andfåddhet, ökad slem och aptitlöshet)
- Sinusvärk eller tryck orsakat av infektion eller polyper
Symtom som kan märkas senare i livet:
- Infertilitet (hos män)
- Upprepad inflammation i bukspottkörteln (pankreatit)
- Andningssvårigheter
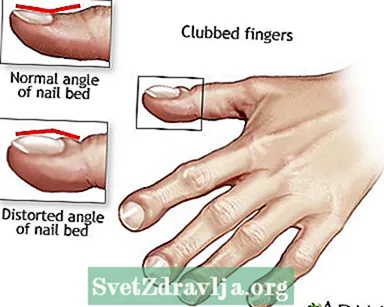
- Klubbade fingrar
Ett blodprov görs för att upptäcka CF. Testet letar efter förändringar i CF-genen. Andra tester som används för att diagnostisera CF inkluderar:
- Immunreaktivt trypsinogen (IRT) test är ett standardtest för nyfödda screening för CF. En hög nivå av IRT föreslår möjlig CF och kräver ytterligare testning.
- Svettkloridtest är standarddiagnostiskt test för CF. En hög saltnivå i personens svett är ett tecken på sjukdomen.
Andra tester som identifierar problem som kan relateras till CF inkluderar:
- Röntgen- eller CT-skanning på bröstet
- Fekalt fett test
- Lungfunktionstester
- Mätning av bukspottkörtelns funktion (avföring pankreas elastas)
- Sekretinstimuleringstest
- Trypsin och chymotrypsin i avföringen
- Övre GI och tunntarmsserier
- Lungkulturer (erhållna genom sputum, bronkoskopi eller svalpinne)
En tidig diagnos av CF och behandlingsplan kan förbättra både överlevnad och livskvalitet. Uppföljning och övervakning är mycket viktigt. När det är möjligt bör vård tas emot på en specialklinik för cystisk fibros. När barn når vuxen ålder ska de övergå till ett specialcenter för cystisk fibros för vuxna.
Behandling för lungproblem inkluderar:
- Antibiotika för att förebygga och behandla lung- och sinusinfektioner. De kan tas i munnen eller ges i venerna eller genom andningsbehandlingar. Personer med CF kan bara ta antibiotika när det behövs eller hela tiden. Doser är ofta högre än normalt.
- Inhalerade läkemedel som hjälper till att öppna luftvägarna.
- Andra läkemedel som ges genom andningsbehandling mot tunn slem och som gör det lättare att hosta upp är DNA-enzymbehandling och högkoncentrerade saltlösningar (hyperton saltlösning).
- Influensavaccin och pneumokockpolysackaridvaccin (PPV) årligen (fråga din vårdgivare).
- Lungtransplantation är ett alternativ i vissa fall.
- Syrebehandling kan behövas eftersom lungsjukdomen blir värre.
Lungproblem behandlas också med terapier för att tunna slem. Detta gör det lättare att hosta slem från lungorna.
Dessa metoder inkluderar:
- Aktivitet eller träning som får dig att andas djupt
- Enheter som används under dagen för att rensa luftvägarna för mycket slem
- Manuell bröstverkan (eller bröstfysioterapi), där en familjemedlem eller en terapeut lätt klappar personens bröst, rygg och område under armarna
Behandling av tarm- och näringsproblem kan innefatta:
- En speciell diet med högt proteininnehåll och kalorier för äldre barn och vuxna
- Bukspottkörtelnzymer som hjälper till att absorbera fetter och protein, som tas med varje måltid
- Vitamintillskott, särskilt vitamin A, D, E och K
- Din leverantör kan ge råd om andra behandlingar om du har mycket hårig avföring
Ivacaftor, lumacaftor, tezacaftor och elexacaftor är läkemedel som behandlar vissa typer av CF.
- De förbättrar funktionen hos en av de defekta generna som orsakar CF.
- Upp till 90% av patienterna med CF och berättigade till en eller flera av dessa läkemedel ensamma eller i kombination.
- Som ett resultat är det mindre ansamling av tjockt slem i lungorna. Andra CF-symtom förbättras också.
Vård och övervakning hemma bör omfatta:
- Undvik rök, damm, smuts, ångor, kemikalier för hushåll, öppen spis och mögel eller mögel.
- Att ge mycket vätska, särskilt till spädbarn och barn i varmt väder, när det finns diarré eller lös avföring eller under extra fysisk aktivitet.
- Träna 2 eller 3 gånger varje vecka. Simning, jogging och cykling är bra alternativ.
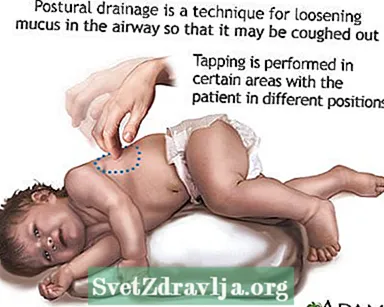
- Rensa eller ta upp slem eller utsöndringar från luftvägarna. Detta måste göras 1 till 4 gånger varje dag. Patienter, familjer och vårdgivare måste lära sig att göra bröstslagverk och postural dränering för att hålla luftvägarna klara.
- Ingen kontakt med andra personer med CF rekommenderas eftersom de kan utbyta infektioner (gäller inte familjemedlemmar).
Du kan lindra sjukdomsstressen genom att gå med i en stödgrupp för cystisk fibros. Att dela med andra som har gemensamma erfarenheter och problem kan hjälpa din familj att inte känna sig ensam.
De flesta barn med CF har god hälsa tills de når vuxen ålder. De kan delta i de flesta aktiviteter och gå i skolan. Många unga vuxna med CF avslutar college eller hittar jobb.
Lungsjukdomen förvärras så småningom till den punkt där personen är funktionshindrad. Idag är den genomsnittliga livslängden för personer med CF som lever till vuxen ålder cirka 44 år.
Döden orsakas oftast av lungkomplikationer.
Den vanligaste komplikationen är kronisk luftvägsinfektion.
Andra komplikationer inkluderar:
- Tarmproblem, såsom gallsten, tarmblockering och rektal prolaps
- Hosta blod
- Kroniskt andningssvikt
- Diabetes
- Infertilitet
- Leversjukdom eller leversvikt, pankreatit, gallcirros
- Undernäring
- Näspolyper och bihåleinflammation
- Osteoporos och artrit
- Lunginflammation som fortsätter att komma tillbaka
- Pneumothorax
- Höger sida hjärtsvikt (cor pulmonale)
- Kolorektal cancer
Ring din leverantör om ett spädbarn eller barn har symtom på CF och upplever:
- Feber, ökad hosta, förändringar i sputum eller blod i sputum, aptitlöshet eller andra tecken på lunginflammation
- Ökad viktminskning
- Oftare tarmrörelser eller avföring som är illaluktande eller har mer slem
- Svullen mage eller ökad uppblåsthet
Ring din leverantör om en person med CF utvecklar nya symtom eller om symtomen förvärras, särskilt allvarliga andningssvårigheter eller hosta upp blod.
CF kan inte förhindras. Screening av personer med en familjehistoria av sjukdomen kan upptäcka CF-genen i många bärare.
CF
- Enteral näring - barn - hanterar problem
- Gastrostomi utfodringsrör - bolus
- Hur man andas när du är andfådd
- Jejunostomy matarrör
- Postural dränering
 Klubbning
Klubbning Postural dränering
Postural dränering Klubbade fingrar
Klubbade fingrar Cystisk fibros
Cystisk fibros
Donaldson SH, Pilewski JM, Griese M, et al. Tezakaftor / ivakaftor hos patienter med cystisk fibros och F508del / F508del-CFTR eller F508del / G551D-CFTR. Am J Respir Crit Care Med. 2018; 197 (2): 214-224. PMID: 28930490 pubmed.ncbi.nlm.nih.gov/28930490/.
Eagan ME, Schechter MS, Voynow JA. Cystisk fibros. I: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, red. Nelson lärobok för pediatrik. 21: e upplagan Philadelphia, PA: Elsevier; 2020: kap 432.
Farrell PM, White TB, Ren CL, et al. Diagnos av cystisk fibros: riktlinjer för konsensus från Cystic Fibrosis Foundation. J Pediatr. 2017; 181S: S4-S15.e1. PMID: 28129811 pubmed.ncbi.nlm.nih.gov/28129811/.
Graeber SY, Dopfer C, Naehrlich L, et al. Effekter av terapi med lumakaftor / ivakaftor på CFTR-funktion hos Phe508del homozygota patienter med cystisk fibros. Am J Respir Crit Care Med. 2018; 197 (11): 1433-1442. PMID: 29327948 pubmed.ncbi.nlm.nih.gov/29327948/.
Grasemann H. Cystisk fibros. I: Goldman L, Schafer AI, red. Goldman-Cecil Medicine. 26: e upplagan Philadelphia, PA: Elsevier; 2020: kap 83.
Rowe SM, Hoover W, Solomon GM, Sorscher EJ. Cystisk fibros. I: Broaddus VC, Mason RJ, Ernst JD, et al, red. Murray och Nadels lärobok för andningsmedicin. 6: e upplagan Philadelphia, PA: Elsevier Saunders; 2016: kap 47.
Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivakaftor hos patienter med cystisk fibros homozygot för phe508del. N Engl J Med. 2017; 377 (21): 2013-2023. PMID: 29099344 pubmed.ncbi.nlm.nih.gov/29099344/.
Vi Rekommenderar Dig Att Läsa

Missa inte medicinska tester